Maladies de stockage
définition
Le terme maladie de stockage englobe un certain nombre de maladies dans lesquelles certaines substances sont déposées dans des organes ou des cellules en raison de processus perturbés dans le métabolisme.
Selon la substance et l'organe, les maladies de stockage peuvent varier considérablement dans leur gravité et leur forme.
Certaines maladies du stockage sont déjà apparentes à la naissance et nécessitent une thérapie immédiate, tandis que d'autres n'apparaissent qu'au cours de la vie.

Quelles sont les maladies de stockage?
-
Maladie du stockage du fer - hémochromatose
-
Maladie du stockage du cuivre - Maladie de Wilson
-
Maladie du stockage des protéines
-
Maladie du stockage du glycogène
-
Maladie du stockage lysosomal
-
Maladie du stockage des esters de cholestérol
-
Maladie du stockage myocardique
-
Maladie neutre du stockage des graisses
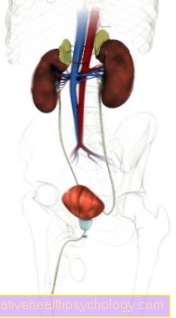
Maladie du stockage du fer
La maladie du stockage du fer, connue dans les milieux spécialisés sous le nom d'hémochromatose, est une maladie métabolique dans laquelle il y a une augmentation des dépôts de fer dans le corps et dans certains organes.
Dans la plupart des cas, la maladie du stockage du fer est une anomalie héréditaire qui conduit à une absorption excessive de fer par le tractus gastro-intestinal.
L'excès de fer absorbé ne peut pas être excrété aussi rapidement qu'il est absorbé et se dépose donc dans divers organes.
Selon l'organe affecté et la quantité de fer, divers symptômes d'hémochromatose et des plaintes peuvent survenir.
Dans de rares cas, la maladie du stockage du fer peut également survenir à la suite d'une autre maladie sous-jacente ou à la suite de transfusions sanguines fréquentes, appelées hémochromatose secondaire.
Le fer supplémentaire stocké dans les organes, qui ne servent normalement pas de réserves de fer, conduit à des processus de remodelage.
Au cours de ces processus de remodelage, une forme de tissu cicatriciel est créée, qui remplace le tissu organique sain et réduit ainsi la fonctionnalité de l'organe.
Souvent, cela affecte principalement les organes producteurs d'hormones dans l'abdomen, comme le foie (le plus souvent) ou le pancréas.
Les organes tels que le cœur, la peau et l'hypophyse font également partie des organes les plus fréquemment endommagés.
L'évolution de la maladie est généralement insidieuse et n'est donc souvent observée qu'à un stade avancé de la maladie.
Les symptômes dépendent de l'étendue des dommages et des organes affectés.
Les symptômes généraux tels que la fatigue et la fatigue sont typiques au début.
Au cours de la maladie, il y a généralement des douleurs articulaires dans les articulations des doigts, en particulier l'index et le majeur, ainsi qu'une coloration brune notable de la peau.
À l'aide de tests sanguins et de biopsies spéciales d'organes individuels, un diagnostic précis peut être posé pour savoir quels organes sont affectés et dans quelle mesure.
Les manifestations d'organes les plus courantes et les plus typiques comprennent, en premier lieu, le foie avec cirrhose du foie, qui est un facteur de risque élevé de développement d'un cancer du foie, et le pancréas avec le développement du diabète sucré.
Les options de traitement de la maladie du stockage du fer se limitent à l'excrétion régulière d'un excès de fer.
Un remède causal n'est pas encore connu.
Tout d'abord, un régime pauvre en fer est recommandé, ainsi qu'une consommation régulière de thé noir, car cela réduit l'absorption du fer dans les intestins.
Si les valeurs de fer sont élevées malgré un régime pauvre en fer, la saignée est la méthode de choix.
Ici, 500 ml de sang sont prélevés sur le patient, de sorte que le fer lié aux cellules sanguines est perdu.
Après avoir atteint le niveau cible de fer dans le sang, une saignée tous les 2-3 mois avec des contrôles de laboratoire étroits est recommandée.
Cette intervention peut souvent être supprimée chez les femmes en âge de procréer, car les saignements menstruels entraînent une perte de fer suffisante.
Comme alternative à la saignée, des médicaments liant le fer sont toujours disponibles, mais ils ne sont utilisés que si la saignée n'est pas possible, par exemple en raison d'une anémie - anémie - ou d'une autre maladie sous-jacente.
Avec un diagnostic précoce et un traitement cohérent, les personnes atteintes de maladie du stockage du fer ont une espérance de vie normale.
Maladie du stockage du cuivre
La maladie du stockage du cuivre, appelée maladie de Wilson, est une maladie métabolique basée sur l'excrétion perturbée du cuivre.
La raison en est un défaut génétique héréditaire dans une protéine qui prépare le cuivre à l'excrétion dans la bile.
S'il y a un défaut ici, le cuivre ne peut plus être excrété en quantité suffisante.
Il s'accumule dans la circulation sanguine et, par conséquent, se dépose dans divers organes.
En règle générale, le cuivre est principalement déposé dans le foie, la cornée, les globules rouges et le cerveau.
En particulier, l'atteinte du foie et du cerveau conduit en combinaison à des symptômes typiques, qui conduisent au diagnostic suspecté d'une maladie du stockage du cuivre.
Les premiers symptômes apparaissent souvent entre 5 et 10 ans, par exemple sous la forme d'une inflammation du foie, appelée hépatite, ou de restrictions neurologiques dues à une fonction hépatique diminuée, comme la somnolence et les mains tremblantes.
À partir de 10 ans, des troubles neurologiques apparaissent généralement, tels que de fins tremblements de la main, une démence, des troubles de la déglutition ou de la parole et des troubles de la marche.
De plus, les dépôts de cuivre peuvent devenir visibles dans l'œil.
Voici un anneau vert-brun dans la cornée.
Le diagnostic de maladie du stockage du cuivre peut être confirmé à l'aide de tests sanguins et urinaires, éventuellement d'une biopsie du foie et de divers tests d'imagerie.
Le traitement primaire avec un diagnostic confirmé d'une maladie du stockage du cuivre consiste en une combinaison d'un régime pauvre en cuivre et de médicaments qui servent à excréter le cuivre, les agents dits chélateurs, par exemple la D-pénicillamine.
Si le traitement est débuté tôt et régulièrement, le pronostic de la maladie est bon.
La seule chose importante est de faire un diagnostic précoce, avant que des dommages aux organes ne puissent survenir en raison des dépôts de cuivre.
Ce qui suit s'applique: toute maladie hépatique incertaine qui n'est pas due à une infection, associée à des troubles du mouvement incertains avant l'âge de 45 ans, doit être clarifiée en ce qui concerne une maladie du stockage du cuivre.
Lire aussi l'article: Le test génétique
Maladie du stockage des protéines
La maladie dite du stockage des protéines n'est pas un tableau clinique reconnu selon l'Organisation mondiale de la santé.
C'est plutôt un concept que le professeur Dr. Lothar Wendt a été développé et publié.
Dans son travail, le professeur Wendt a poursuivi une approche alternative pour expliquer les maladies courantes dans notre société, opposant la vision de la médecine traditionnelle à la question du «pourquoi».
Un exemple typique de cette approche peut être illustré par la maladie courante du diabète.
Dans le diabète sucré de type 2, le taux de sucre dans le sang est très élevé.
Cette augmentation de la glycémie entraîne des dommages dans tout le corps avec de graves complications.
L'approche médicale conventionnelle consiste donc à abaisser systématiquement le taux de sucre dans le sang afin d'éviter d'autres dommages.
Le professeur Wendt, quant à lui, demande dans son concept de travail pourquoi cette augmentation de la glycémie se produit et si la raison en est une compensation.
Ici, il avance la théorie selon laquelle les dépôts de protéines dans les parois des vaisseaux sanguins provoquent leur épaississement.
Le professeur Wendt explique que l'augmentation du taux de sucre dans le sang est une réaction à l'épaississement des parois des vaisseaux sanguins afin de transporter une quantité suffisante de sucre dans la cellule malgré la résistance accrue et le chemin de diffusion plus long.
Selon Wendt, ce n'est pas le sucre qui est le facteur causant la maladie, mais la protéine et finalement le terme de diabète sont trompeurs.
Le terme augmentation de la glycémie à la suite d'une maladie causale du stockage des protéines serait plus approprié selon son concept.
À l'heure actuelle, cependant, il y a un manque d'études fondées sur des preuves qui soutiendraient cette approche explicative et le concept de la maladie.
Ce n'est que dans le traitement de l'arthrose qu'il y a déjà des personnes touchées dans certains groupes d'entraide qui déclarent avoir soulagé ou même éliminé l'arthrose grâce à une thérapie ciblée de dégradation des protéines.
Il faut cependant noter qu'il s'agit de valeurs empiriques individuelles sans groupe de référence, qui n'ont réussi que si le traitement a été démarré à un stade très précoce de la maladie.
Des professeurs éminents de divers départements ne voient, compte tenu de la situation actuelle de l'étude, aucune preuve de l'exactitude du concept de maladie du stockage des protéines par le professeur Wendt.
Maladie du stockage du glycogène
Dans les maladies de stockage du glycogène, un défaut génétique héréditaire entraîne un dépôt excessif de glycogène dans le corps.
Le glycogène est également connu sous le nom d'amidon hépatique.
Il s'agit d'une molécule de glucose longue et multi-ramifiée, qui est notamment stockée dans le foie et sert de fournisseur du sucre porteur d'énergie.
Il existe au total neuf formes différentes de maladie du stockage du glycogène, dont chacune est basée sur un défaut génétique différent et conduit au dépôt de glycogène dans différents organes.
Les formes les plus courantes comprennent la maladie de stockage du glycogène de type I, la maladie de von Gierke, la maladie de stockage du glycogène de type II, la maladie de Pompe et la maladie de stockage du glycogène de type V, la maladie de McArdle.
Les différentes formes diffèrent à la fois dans leurs symptômes et dans l'apparition de la maladie.
La maladie du stockage du glycogène de type I est généralement perceptible par une hypertrophie du foie et un abdomen distendu, en plus il y a souvent des convulsions et une tendance à saigner.
Dans la maladie de stockage du glycogène de type II, une fonte musculaire sur tout le corps et une langue surdimensionnée sont particulièrement visibles.
Dans la maladie de stockage du glycogène de type V, également, une fonte musculaire généralisée se produit, mais en combinaison avec des douleurs musculaires et des crampes après l'effort.
Le traitement des maladies de stockage du glycogène dépend du type de maladie et de sa gravité.
Maladie du stockage lysosomal
Le terme maladie de stockage lysosomal englobe un grand groupe de maladies qui sont basées sur un défaut génétique dans les lysosomes.
Les lysosomes sont un groupe de cellules du corps humain qui agissent comme l'estomac ou la poubelle de cellules.
Tous les composants cellulaires en excès et les déchets de la cellule sont décomposés dans les lysosomes.
Si les lysosomes sont défectueux, ces déchets cellulaires s'accumulent, qui se déposent ensuite à la fois dans la cellule et dans d'autres organes.
45 maladies sont actuellement classées dans le groupe des maladies du stockage lysosomal.
La plupart des maladies sont des variantes très rares de la maladie du stockage.
Les formes les plus courantes de maladie du stockage lysosomal sont la maladie de Gaucher et la maladie de Fabry.
En savoir plus sur le sujet: Maladie de Fabry
Dans la maladie de Gaucher, les processus de dégradation perturbés conduisent à une accumulation de graisses dans les cellules et autres organes.
Les symptômes varient considérablement en raison du risque d'affecter tout le corps.
L'élargissement du foie et de la rate, les troubles du système hématogène et les convulsions sont typiques.
La maladie est souvent perceptible dans la petite enfance en raison d'un trouble de l'alimentation.
La maladie de Fabry, en revanche, est significativement plus rare que la maladie de Gaucher et affecte principalement les garçons en raison de son héritage.
Les symptômes de la maladie de Fabry comprennent initialement des brûlures de douleur dans les doigts, des troubles gastro-intestinaux et une opacité cornéenne.
Plus tard, le cœur peut être infecté par une insuffisance cardiaque et des accidents vasculaires cérébraux.
En savoir plus sur le sujet ici: Maladie de Gaucher.
Maladie du stockage des esters de cholestérol
La maladie du stockage des esters de cholestérol appartient au groupe des maladies du stockage lysosomal, qui est une maladie métabolique héréditaire rare.
La maladie des esters de cholestérol est basée sur un défaut de la lipase acide lysosomale, qui décompose normalement les graisses telles que les esters de cholestérol et les triacylglycérides.
La dégradation réduite de ces graisses conduit à une accumulation de graisses dans la cellule et par conséquent également dans la circulation de l'organisme.
Cette maladie ne cause pas de plaintes pendant longtemps, seule l'élargissement réactif du foie peut entraîner une sensation de pression dans l'abdomen supérieur droit, des nausées ou des ballonnements.
Les tests sanguins montrent des valeurs sanguines accrues pour le cholestérol et les lipides, ainsi que des valeurs diminuées pour les bonnes graisses (HDL).
Une stéatose hépatique peut apparaître lors de l'examen échographique du haut de l'abdomen.
Le traitement de la maladie du stockage des esters de cholestérol a lieu avec des médicaments en inhibant l'absorption du cholestérol avec la colestyramine ou l'ézétimibe et en abaissant en outre les taux de lipides sanguins avec des statines telles que la simvastatine.
Maladie du stockage myocardique
Dans la maladie de stockage myocardique, des produits de dégradation se déposent dans les parois du cœur, ce qui peut gravement restreindre les performances du cœur et la fonction de pompage.
Deux maladies de stockage différentes peuvent conduire à ces dépôts dans les parois du cœur: la maladie rare et héréditaire du stockage lysosomal de Fabry et la soi-disant amylose.
Dans la maladie de Fabry, une anomalie génétique héréditaire entraîne une réduction de la dégradation des produits métaboliques, qui se déposent, entre autres, dans les parois cardiaques et peuvent entraîner de graves dommages.
L'amylose, en revanche, peut être héréditaire et acquise au cours de la vie.
Avec ce tableau clinique, il existe également des dépôts de produits métaboliques anormalement modifiés qui, en plus d'autres organes, s'accumulent principalement dans le cœur et limitent ici gravement la fonction du cœur.
Une maladie de stockage myocardique devient perceptible au début par des symptômes généraux tels que faiblesse et fatigue.
Au fil du temps, il y a un essoufflement croissant après l'exercice et à un moment donné également au repos.
L'eau dans les poumons, dans l'abdomen, dans les jambes et dans le péricarde sont des effets secondaires typiques à mesure que la maladie progresse.
Des procédures d'imagerie et une biopsie du muscle cardiaque sont nécessaires pour un diagnostic clair de la maladie du stockage myocardique.
La thérapie ultérieure est alors basée sur la maladie sous-jacente qui l'a provoquée.
Maladie neutre du stockage des graisses
Les maladies neutres de stockage de graisse sont des maladies très rares dans lesquelles la dégradation et le stockage d'une graisse, appelée triglycéride, sont défectueux.
À ce jour, seuls 50 cas de maladies du stockage des graisses neutres ont été décrits dans le monde.
Comme pour la plupart des maladies de stockage, la cause du défaut génétique est également héréditaire dans la maladie de stockage de graisse neutre.
La maladie est souvent constatée dans la petite enfance en raison d'un trouble du développement.
La majorité des personnes atteintes développent une hypertrophie du foie avec un dysfonctionnement hépatique associé, ainsi que des problèmes oculaires et une perte auditive.
Une fonte musculaire et des troubles de la marche peuvent survenir à un âge avancé.


.jpg)