Maladies génétiques
définition
Une maladie génétique ou une maladie héréditaire est une maladie causée par un ou plusieurs gènes de la personne concernée. L'ADN agit ici comme un déclencheur direct de la maladie. Pour la plupart des maladies génétiques, les emplacements des gènes responsables sont connus. En cas de suspicion de maladie génétique, le diagnostic respectif peut donc être posé par un examen génétique.
D'autre part, il existe également un certain nombre de maladies dont la survenue a une influence génétique ou est discutée, comme le diabète sucré («diabète»), l'ostéoporose ou la dépression. Ce sont des soi-disant dispositions, c'est-à-dire une probabilité accrue de certaines maladies. Les dispositions doivent être distinguées des maladies héréditaires.

Ce sont des maladies héréditaires courantes
En termes absolus, les maladies héréditaires ne sont pas courantes, mais les maladies héréditaires énumérées ici se produisent fréquemment par rapport à d'autres maladies de cause génétique.
-
Syndrome de Marfan
-
L'anémie falciforme
-
Hémophilie (hémophilie A ou B)
-
Mutation du facteur V Leiden et résistance à l'APC qui en résulte
-
Faiblesse rouge vert
-
Déficit en glucose-6-phosphate déshydrogénase (déficit en G6PD)
-
Polydactylie («doigts multiples», également possible comme symptôme dans d'autres maladies)
-
Trisomie 21 (syndrome de Down)
-
Chorée Huntington
causes
Les maladies héréditaires sont extrêmement diverses dans leur apparence. Ils n'ont fondamentalement qu'une chose en commun: la cause de chacun d'eux réside dans l'ADN, c'est-à-dire dans le matériel génétique de la personne concernée. Divers changements peuvent se produire ici, tels que des mutations (échange d'informations ADN) ou des suppressions (manque de certains matériels génétiques).
Une grande quantité d'informations est codée dans le matériel génétique, comme les «plans» pour divers composants qui sont importants pour le fonctionnement d'une cellule corporelle. Il peut s'agir par exemple d'enzymes, de canaux électrolytiques ou de substances messagères. Ces plus petits éléments sont alors lus incorrectement ou pas du tout à partir de l'ADN, qui manque alors dans le système sophistiqué du corps. Une information génétique erronée ou manquante entraîne donc certains dysfonctionnements dans l'organisme. Celles-ci provoquent alors des symptômes selon le système fonctionnel dans lequel un élément est désormais manquant.
Découvrez tout sur le sujet ici: Le test génétique.
C'est ainsi que les maladies héréditaires sont héritées
Chaque maladie héréditaire est héréditaire de manière monogénétique ou polygénique: cela signifie qu'il y a un ou plusieurs emplacements génétiques qui doivent être modifiés pour conduire à une maladie.
De plus, les traits génétiques peuvent toujours être hérités de manière dominante ou récessive: récessif signifie que les gènes paternels et maternels doivent être prédisposés à cette maladie héréditaire particulière. Dans le cas de l'hérédité dominante, un seul changement (c'est-à-dire un parent) suffit pour déclencher la maladie. Il s'ensuit qu'avec les maladies héréditaires dominantes, les personnes porteuses tomberont également malades - alors qu'avec un héritage récessif, on ne sait généralement même pas qu'il existe une prédisposition génétique correspondante.
Il existe également des maladies héréditaires des chromosomes sexuels, comme l'hémophilie ou la cécité rouge-vert. Les installations pour cela sont généralement sur le chromosome X, car le chromosome Y est globalement très petit et peut généralement stocker peu d'informations génétiques. On parle donc de maladies héréditaires liées à l'X. Ceux-ci affectent généralement beaucoup plus d'hommes que de femmes, car les femmes peuvent compenser toute information incorrecte sur le chromosome X avec la seconde.
La manière exacte dont une maladie génétique est héritée est généralement facile à rechercher si vous êtes intéressé.
Tests avant la naissance
En principe, le matériel génétique de l'enfant peut déjà être examiné dans l'utérus pour toutes les maladies héréditaires dont les localisations génétiques causales sont connues. Cependant, les analyses génétiques prennent du temps, de sorte que, généralement, seule la localisation du gène suspecté est analysée - pour cela, à son tour, il doit y avoir une suspicion justifiée de maladie génétique.
Pour un tel examen, le matériel génétique peut ensuite être prélevé dans le liquide amniotique ou le placenta et utilisé pour l'analyse.
Cependant, il faut toujours garder à l'esprit que tout diagnostic invasif comporte également un risque pour la vie de l'enfant à naître. Ces crevaisons doivent donc être pesées individuellement dans chaque cas.
Il existe également des mesures qui peuvent indiquer une maladie génétique, comme la mesure de la transparence nucale en tant que signe de trisomie 21. Ces méthodes ne sont pas dangereuses pour l'enfant à naître, mais ne peuvent offrir une certitude absolue sur la présence d'une maladie génétique. Alors là aussi, une opération doit être soigneusement envisagée.
Trisomie 21
La cause de la trisomie 21 est le chromosome 21, qui n'est pas présent deux fois mais trois fois chez les personnes atteintes. Cette variante de l'ADN est créée lorsque les chromosomes sont répartis dans les cellules germinales parentales, c'est-à-dire les spermatozoïdes ou les ovules. Il s'agit donc d'une "erreur de distribution" et non d'une modification du matériel génétique réel. Cela explique pourquoi la trisomie 21 peut survenir spontanément dans toutes les familles et pourquoi la probabilité d'avoir un enfant trisomique est la même dans toutes les familles. À proprement parler, la trisomie 21 - comme les autres trisomies - ne doit pas être considérée comme une maladie héréditaire au vrai sens du terme. Néanmoins, la trisomie 21 est la maladie liée à l'ADN la plus courante chez les nouveau-nés.
Les caractéristiques de l'ensemble modifié de chromosomes dans le syndrome de Down peuvent déjà être observées chez l'enfant à naître dans l'utérus: des retards de croissance et des défauts peuvent entraîner, entre autres, un crâne trop petit, des os courts de la cuisse et du bras et des malformations cardiaques. Une grande quantité de liquide amniotique peut également être une indication de la trisomie 21, car les enfants à naître atteints boivent ou avalent relativement peu de liquide amniotique. Cependant, aucune de ces caractéristiques n'est un signe définitif du syndrome de Down!
En plus des signes de retard de croissance mentionnés, les enfants trisomiques présentent souvent un retard de développement, par exemple dans les domaines du langage et de la motricité. Les personnes atteintes du syndrome de Down font souvent preuve de compétences sociales remarquables, tandis que l'intelligence reste souvent inférieure à la moyenne. Cependant, les personnes touchées diffèrent considérablement dans ces caractéristiques; il n'est pas rare qu'elles obtiennent leur diplôme après avoir reçu un bon soutien.
Plus tard dans la vie, les personnes atteintes de trisomie 21 ont un risque accru d'être diagnostiquées avec certaines maladies. Ceux-ci incluent la maladie d'Alzheimer, l'épilepsie et le cancer, en particulier la leucémie. Néanmoins, l'espérance de vie des personnes atteintes du syndrome de Down continue d'augmenter: entre-temps, les personnes atteintes atteignent souvent l'âge de 60 ou 70 ans.
Vous pouvez trouver plus d'informations sur notre site Web Syndrome de bas
Déficit en alpha-1 antitrypsine
La carence en alpha-1 antitrypsine peut prendre différentes formes et formes, selon les caractéristiques génétiques exactes de la personne touchée. Cela signifie que toutes les carences en alpha-1 antitrypsine ne provoquent pas de symptômes. Dans ce qui suit, seul le type cliniquement évident (PiZZ) de cette maladie génétiquement déterminée sera discuté.
Le défaut enzymatique présent dans cette maladie provoque la dégradation et le remodelage des éléments constitutifs des tissus organiques chez les personnes atteintes. De plus, les protéines défectueuses sont filtrées hors du sang par le foie et s'y accumulent. Cela peut entraîner une inflammation du foie (hépatite), une cirrhose ou un cancer du foie. Les voies respiratoires des poumons deviennent instables en raison du manque de tissu stable et elles s'effondrent plus rapidement: Le tableau clinique de la BPCO (maladie pulmonaire obstructive chronique) se développe. Ce tableau clinique est souvent le premier symptôme d'un déficit en alpha-1 antitrypsine, de sorte que toute personne atteinte de BPCO à un plus jeune âge doit être contrôlée pour un déficit en alpha-1 antitrypsine.
Si la maladie persiste depuis longtemps, les poumons peuvent gonfler excessivement, car l'air que vous respirez ne peut pas être correctement expiré par les voies respiratoires instables et s'accumule dans les poumons. En tant que thérapie, en plus d'éviter systématiquement le tabagisme et des vaccinations régulières pour prévenir les maladies respiratoires, des mesures médicales doivent également être prises: l'alpha-1-antitrypsine manquante peut être administrée par voie intraveineuse afin d'atténuer les symptômes autant que possible et d'arrêter l'évolution de la maladie.
Vous pouvez trouver plus d'informations sur notre site Web Déficit en alpha-1 antitrypsine
hémophilie
Le groupe de l'hémophilie est également appelé familièrement «hémophilie», car ce terme décrit très précisément le principal symptôme de cette maladie héréditaire: les personnes touchées saignent plus longtemps et, selon la gravité de la maladie, plus souvent que saines.
Le saignement est généralement arrêté par ce que l'on appelle la cascade de coagulation, une voie de signalisation endogène qui empêche une perte de sang excessive. Dans ce système de coagulation, 13 facteurs jouent un rôle, qui s'activent les uns après les autres. Cela peut être imaginé comme une série de dominos: si vous frappez une pierre (facteur de coagulation), cela active la suivante, et ainsi de suite. À la fin de ce chemin de signal ou des dominos, il y a une coagulation sanguine. Avec l'hémophilie, il manque un certain facteur - selon le sous-type spécifique de la maladie: la réaction en chaîne s'interrompt ici.
Le traitement de la maladie peut être effectué en déterminant le facteur manquant et en l'ajoutant de l'extérieur. Les personnes concernées doivent donc s'injecter régulièrement une préparation avec ce facteur de coagulation afin que le reste de la réaction en chaîne puisse avoir lieu.
Vous pouvez trouver plus d'informations sur notre site Web Maladie du sang
Fibrose kystique
Dans la maladie génétique la fibrose kystique - également appelée fibrose kystique - il y a une production défectueuse de canaux ioniques, plus précisément de canaux chlorures. En conséquence, la composition des sécrétions corporelles (par exemple, la sueur, les sécrétions des voies respiratoires et du pancréas) des personnes affectées est modifiée: Étant donné que le manque de chlorure signifie que moins d'eau est aspirée dans le conduit de la glande respective, la sécrétion est relativement visqueuse.
En conséquence, les symptômes se développent généralement dans le tube digestif, car la sécrétion avec les enzymes digestives ne peut pas bien s'écouler du pancréas dans l'intestin et endommage ainsi le pancréas lui-même. De plus, les troubles digestifs tels que les selles graisseuses, la diarrhée et le faible poids corporel qui en résulte sont courants.
Le deuxième grand groupe de symptômes se développe généralement dans les poumons: puisque le mucus qui se produit naturellement dans les poumons est plus visqueux que chez les personnes en bonne santé, il est plus difficile de l'éliminer des cils. Cela peut entraîner une toux chronique et des blocages des bronches (bronchectasie). La plus grande quantité de sécrétion pulmonaire fournit également un bon environnement pour la croissance des bactéries, ce qui entraîne des infections respiratoires fréquentes et une pneumonie.
La fibrose kystique est traitée de manière symptomatique avec des expectorants, des enzymes digestives et des antibiotiques pour les infections.
Vous pouvez en savoir plus sur notre site Web Fibrose kystique
Facteur V Leiden et résistance APC
Une mutation du facteur V Leiden implique une modification de l'information génétique qui peut entraîner une augmentation de la coagulation sanguine. La raison en est le facteur V dans la soi-disant cascade de coagulation du corps: ce chemin de signal garantit qu'en cas de blessure, la plaie est fermée par les propres «protéines adhésives» du corps (fibrine). Il y a 13 facteurs dans ce chemin de signalisation, qui sont nommés avec des chiffres romains (cela signifie «Facteur 5 souffrance»!). Le facteur V a un effet bénéfique sur la formation d'un bouchon de fibrine, mais peut également être inhibé par la protéine C dite activée (APC en abrégé). Cela joue un rôle important dans la régulation de cette voie de signalisation et dans la prévention de la coagulation sanguine excessive.
Le facteur V muté est présent chez les individus affectés mais ne répond pas à l'APC. Le corps ne dispose pas d'un "dispositif de sécurité" important à ce stade pour empêcher la coagulation du sang sans raison, ce qui peut même bloquer les vaisseaux et provoquer ainsi des troubles circulatoires.
Statistiquement parlant, les personnes atteintes d'une mutation du facteur V Leiden sont plus susceptibles de subir un événement thrombotique (c'est-à-dire une thrombose ou une embolie pulmonaire), même sans antécédents de facteurs de risque typiques. En termes techniques, on parle aussi de «thrombophilie», c'est-à-dire de tendance à coaguler.
Vous pouvez en savoir plus sur notre site Web Facteur V Leiden
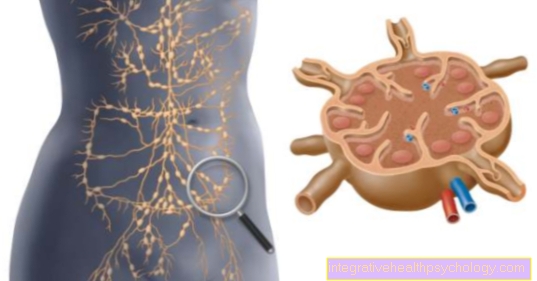
Maladie de Gaucher
Dans la maladie de Gaucher, la modification des informations ADN provoque un défaut dans une enzyme impliquée dans le métabolisme lipidique, plus précisément la glucocérébrosidase: cela aide à décomposer les anciens composants cellulaires. En cas de défaut, il peut y avoir une diminution de la fonctionnalité ou même une perte de fonctionnalité, et en conséquence les symptômes apparaissent dans l'enfance ou le jeune adulte.
Les symptômes de la maladie de Gaucher sont en grande partie dus à une hypertrophie du foie et de la rate, dont le corps tente de compenser le manque d'enzymes. Cela augmente la dégradation de tous les composants sanguins, qui peuvent être reconnus dans la formule sanguine et utilisés comme indicateur de diagnostic avec le foie et la rate hypertrophiés.
L'enzyme glucocérébrosidase manquante peut être utilisée en thérapeutique comme médicament. Le pronostic et l'évolution de la maladie de Gaucher dépendent en grande partie de la gravité de la perte de fonction de l'enzyme.
Pour plus d'informations, lisez ici: Maladie de Gaucher.
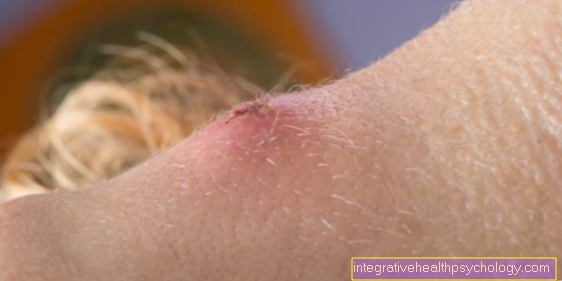
Maladie d'Osler
La maladie d'Osler est une maladie héréditaire qui se caractérise par une forte vasodilatation. En principe, cette expansion des vaisseaux peut se produire n'importe où, à la fois sur la peau et sur les organes internes. Les parois des vaisseaux élargis sont relativement minces et se déchirent facilement. En conséquence, les zones touchées saignent rapidement.
La vasodilatation se produit particulièrement fréquemment sur le visage et dans la muqueuse nasale, de sorte que les personnes touchées se plaignent généralement de saignements de nez fréquents et de petits saignements tachetés sur le visage.
En cas de suspicion de maladie d'Osler, des diagnostics appropriés doivent être effectués, car la vasodilatation peut également survenir dans des organes vitaux ou des organes avec une bonne irrigation sanguine, tels que les poumons, le cerveau ou le foie, dans lesquels l'hémorragie d'un vaisseau rompu est dangereuse.
Vous pouvez en savoir plus sur ce sujet sur notre site Web Maladie d'Osler
Maladie de Recklinghausen
La neurofibromatose de type 1 - ou maladie de Recklinghausen - est une maladie génétique dans laquelle les personnes touchées développent souvent des tumeurs sur les cellules de la couverture nerveuse. Les tumeurs qui se développent peuvent être à la fois bénignes et malignes et apparaître à un jeune âge.
Les tumeurs typiques, cependant, sont des neurofibromes bénins: il s'agit de cellules qui enveloppent et isolent le nerf comme un câble électrique, ainsi que le tissu conjonctif environnant. Ce sont des tumeurs bénignes, c'est-à-dire non propagées et à croissance lente.
Cependant, la chirurgie pour enlever les neurofibromes peut être difficile, car ils sont souvent fermement attachés au nerf et le nerf correspondant doit alors être retiré. Néanmoins, c'est la seule option de traitement pour le neurofibrome symptomatique, car le traitement causal de cette maladie héréditaire n'est pas possible.
Vous pouvez en savoir plus sur ce sujet sur notre site Web Neurofibromatose de type 1
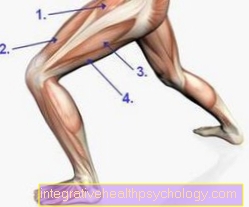
Dystrophie musculaire
Le terme dystrophie musculaire décrit un groupe de maladies héréditaires dans lesquelles certains composants musculaires ne peuvent pas ou ne peuvent pas être assemblés correctement par les cellules du corps. En conséquence, les personnes touchées développent généralement une faiblesse musculaire dès l'enfance et l'adolescence, ce qui peut entraîner une perte de masse musculaire, des restrictions de mouvement et même des incapacités physiques.
Si la présence d'une dystrophie musculaire est suspectée, les valeurs sanguines doivent d'abord être déterminées. Si les valeurs correspondent au diagnostic suspecté, une biopsie musculaire peut toujours être effectuée: un petit échantillon de tissu est prélevé sur le muscle, qui est ensuite examiné au microscope pour détecter des défauts cellulaires. Un examen génétique est également possible pour établir le diagnostic, car les localisations génétiques correspondantes sont généralement connues pour les différentes formes de dystrophie musculaire et devraient être modifiées. Un traitement causal des dystrophies musculaires n'est pas connu.
Vous pouvez en savoir plus sur ce sujet sur notre site Web Dystrophie musculaire
Xeroderma pigmentosum
Xeroderma pigmentosum est une maladie héréditaire rare dans laquelle certaines enzymes de la peau de la personne affectée ne fonctionnent pas. Ces enzymes s'occupent normalement de la réparation de l'ADN, qui peut être endommagé par la lumière du soleil ou la lumière UVB contenue. Les dommages causés par les UVB peuvent provoquer un cancer de la peau chez les personnes touchées ainsi que chez toutes les autres personnes, mais avec Xeroderma Pigmentosum, le processus est accéléré par l'absence de mécanismes de réparation. En conséquence, les personnes touchées développent des formes sévères de cancer de la peau pendant l'enfance et l'adolescence et après une courte exposition au soleil.
Une thérapie causale n'est pas encore possible. Les personnes touchées doivent éviter la lumière du soleil toute leur vie, c'est pourquoi le surnom «d'enfants au clair de lune» s'est imposé pour les personnes touchées (parfois très jeunes). De plus, ces personnes doivent être supervisées par un dermatologue pour un dépistage régulier du cancer de la peau afin d'éliminer immédiatement le nouveau cancer de la peau. Si ces mesures sont strictement suivies, l'espérance de vie d'une personne atteinte de xeroderma pigmentosum est à peu près la même que celle d'une personne non atteinte.
Vous pouvez en savoir plus sur cette maladie sur notre site Web Xeroderma pigmentosum
Syndrome de Lynch
Le syndrome de Lynch est une modification de l'ADN qui provoque une enzyme défectueuse dans les cellules du corps.Chez les personnes touchées, un certain mécanisme est défectueux, qui est autrement censé protéger les cellules de la dégénérescence, c'est-à-dire une croissance incontrôlée - les personnes atteintes du syndrome de Lynch ont donc un risque considérablement accru de développer un cancer.
Le cancer du côlon se produit souvent ici parce que les cellules se divisent naturellement souvent ici de toute façon et les erreurs dans la programmation de la croissance et de la mort d'une cellule deviennent plus rapidement perceptibles. Les personnes atteintes développent souvent une tumeur dans le gros intestin à un âge inhabituellement jeune, c'est-à-dire avant l'âge de 50 ans, qui est alors appelée HNPCC (cancer héréditaire du côlon non polypeux). Cependant, tous ceux qui ont la constitution génétique du syndrome de Lynch ne développeront pas un cancer du côlon. D'autre part, d'autres organes peuvent également développer une tumeur, puisque les prédispositions génétiques qui favorisent le développement d'une tumeur sont présentes dans toutes les cellules du corps. Des contrôles réguliers et des examens préventifs sont donc nécessaires pour les personnes atteintes du syndrome de Lynch afin de traiter adéquatement les tumeurs qui se développent à un stade précoce.
Vous pouvez en savoir plus sur ce sujet sur notre site Web Syndrome de Lynch